A Practical Guide for Single Domain Antibody Production
Antibodies are biological protein molecules produced by immune B cells stimulated by antigens and can bind to antigens specifically. Due to their ability to bind antigens with high specificity and high affinity, antibodies are widely used in academic research, disease diagnosis and medical drugs.
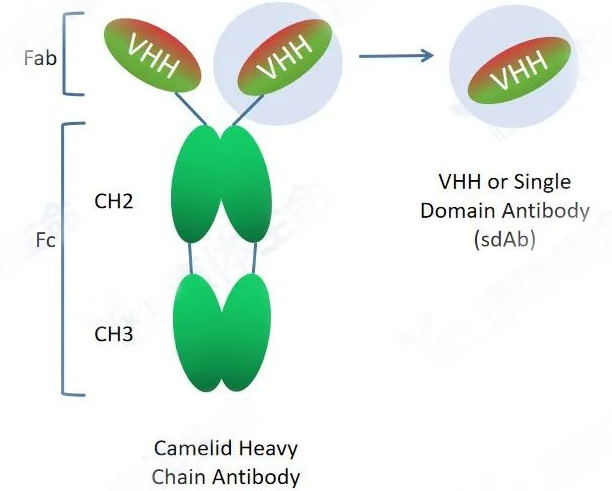
The conventional antibody (IgG) is a protein complex composed of two identical heavy chains and two identical light chains. The light chain contains one variable region and one constant region, named VL and CL, respectively. The heavy chain contains one variable region and three constant regions, named VH, CH1, CH2 and CH3, respectively. The VH region and the VL region together constitute the smallest antigen recognition unit of conventional antibodies, and the sequence difference of the antibody variable region determines that it can specifically recognize and bind different antigens. The CL and CH regions are relatively conserved. The CH2 and CH3 regions play an important role in the recruitment of immune cells to perform ADCC and CDC functions.
Heavy chain antibody is a special antibody composed of only two heavy chains naturally occurring in camelids and cartilaginous fish and includes a variable domain of heavy chain (VHH, Variable domain of heavy chain antibody) and two conventional CH2 and CH3 regions. The CH1 region is naturally absent. Heavy chain antibodies bind to antigens through the variable region (VHH) on the heavy chain, which can exist independently and stably in vitro. VHH is also named as camelid single domain antibody (SdAb) or nanobody. VHH crystals are about 2.5nm in diameter and 4nm in length, and its molecular weight is only 1/10 (about 12-15KD) of conventional intact antibodies (about 150KD), but they still keep complete antigen recognition capabilities with high affinity.
Compared with conventional antibodies, nanobodies are smaller, more hydrophilic and can be mass-produced by recombinant expression in E. coli or yeast, effectively avoiding the batch-to-batch production variation of conventional antibodies. Due to these characteristics, nanobodies have a series of advantages in new drug discovery, showing great potential:
It can bind to protein conformational epitopes that cannot be recognized by conventional antibodies, and has higher specificity to the target.
Better tissue penetration
Higher stability
Suitable for industrialized large-scale production
Easier to modify and optimize
Easier to humanize
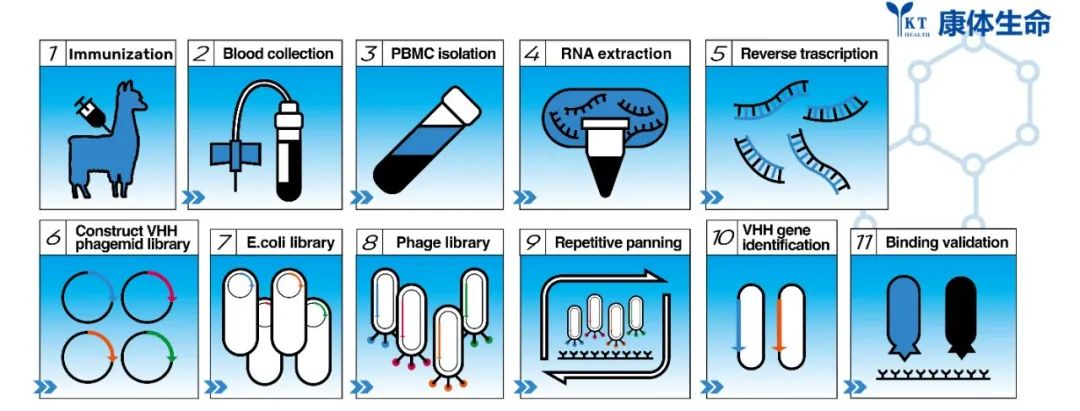
Due to these characteristics of nanobodies, more and more research institutions and drug manufacturers are paying attention to and trying to use nanobodies in different scenarios. The development of nanobodies is different from the traditional method of preparing monoclonal antibodies through hybridomas. Nanobodies can be discovered by immunizing alpacas with antigen, constructing phage libraries using isolated PBMC after successful immunization, and screening by phage display and phage ELISA. Positive single domain antibody candidates are then expressed and purified to further verify whether they can bind to the antigen specifically or not. Here, we will describe in detail the process of discovering nanobodies in our lab, as well as the key points and operating skills. Each step has been optimized and summarized by Alpa Life for your reference.
General experimental process of single domain antibody development and discovery:
The experimental process mainly includes alpaca immunization, phage library construction, VHH screening, VHH candidate expression, purification, and verification. After antigen immunization in alpacas,the specific antibodies are matured in vivo by the action of the immune system. B lymphocytes are then isolated for extraction of total RNA. cDNA is obtained by reverse transcription using RNA as template. cDNA is then used as template for nested PCR to obtain diverse VHH encoding frame DNA fragments. The VHH DNA fragments are then enzyme digested and linked to phagemids to form phage libraries. Nanobodies are screened by phage display and screening technology, and the positive VHH candidates were further verified by Elisa or SPR analysis.
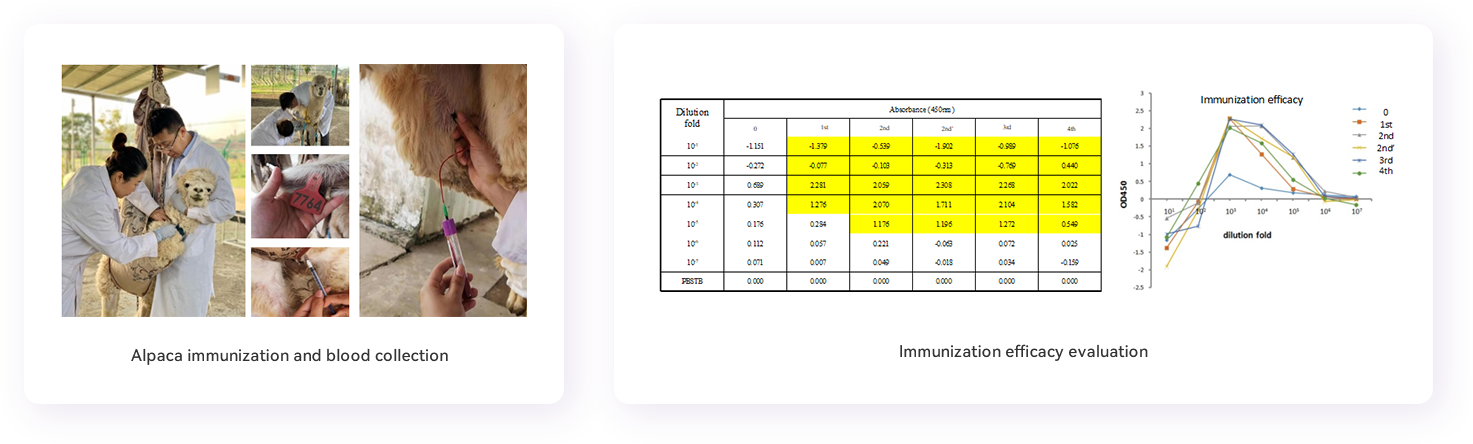
1. Antigen preparation: Antigen quality including purity and bioactivity should be confirmed before immunization. Multiple freeze-thaw cycles should be avoided to mimimize denaturation. An alpaca can be immunized with 1-3 protein antigens at the same time. Each antigen for each immunization injection should be kept at about 0.5mg and the total antigen volume should be less than 1.5 mL. The volume of antigen and adjuvant emulsified would be 1:1 before immunization.
2. Alpaca immunization: Emulsified antigen is injected subcutaneously at multiple locations near the neck draining lymph nodes (neck base near the bow lymph node) of the alpaca. Each location is injected with about 0.4 mL of the emulsified antigen. Immunized alpaca will be observed for half an hour to confirm that the animal is in good condition and has no side symptoms. Immunizations are usually performed for at least 4 times at bi-weekly intervals.
3. Serum separation: Before each immunization, serum should be collected for immune conversion evaluation. 5 mL of blood will be taken each time, centrifuged at 4000 x g for 30 minutes in a pre-cooled 25 °C centrifuge. The upper serum fraction will be separated for storage at -20°C for subsequent immune conversion evaluation.
4. Blood sampling: collect 50 mL of blood from jugular vein of the alpaca in EDTA-coated blood collection tubes to avoid coagulation 5-7 days after the fourth immunization.
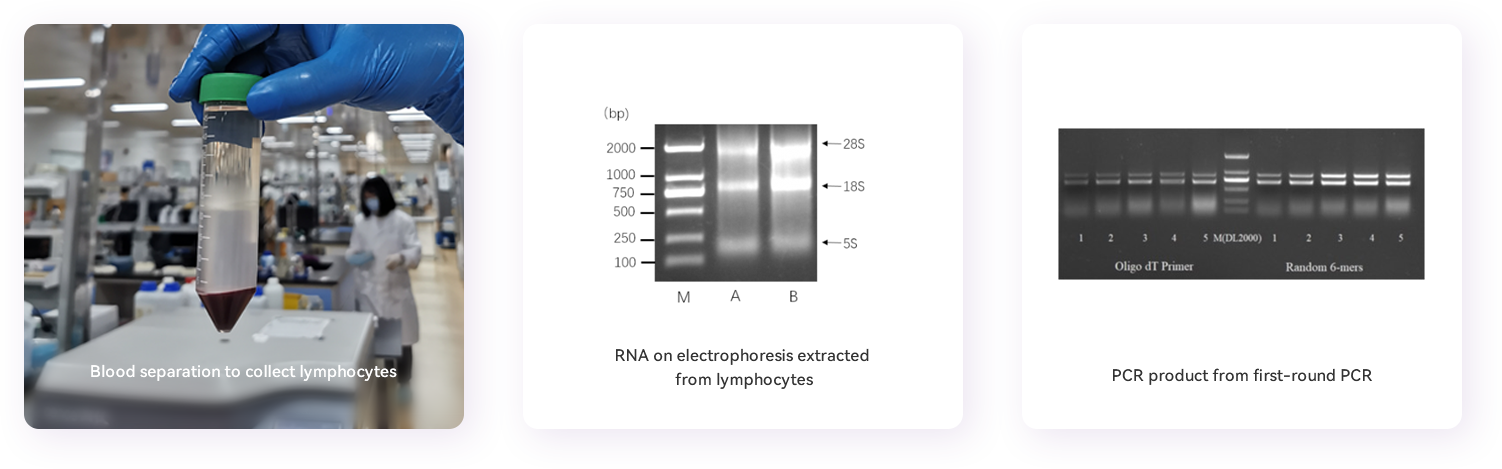
5. lymphocytes isolation: 15 mL of cell separation solution is pre-filled to a 50 mL centrifuge tube, and then 15 mL of blood will be slowly added on top of the the cell separation solution to prevent mixing. Centrifuge at 400 x g for 30 minutes at 25°C. The upper plasma sample will be stored in a new centrifuge tube at -20°C for immune conversion evaluation. The cotton-like layer containing the lymphocytes will be transferred carefully with a pipette into a new 50 mL centrifuge tube. 10 mL of PBS buffer will then be added to the tube to wash the cells and centrifuge at 400 x g for 20 minutes at 25°C. Discard the supernatant, 5 mL of PBS will be added at room temperature, mix gently, count the number of cells using a hemocytometer, and then centrifuge at 400 x g for 20 minutes at 25°C. Discard the supernatant, RNAiso Plus solution will be added to the isolated lymphocytes according to the number of cells to obtain 107/mL cell lysate and stored at -80°C for later RNA extraction.
Tips:
1. The selection of alpacas and the antigens are key points for successful immunization. Choose a healthy, strong, moderately sized and non-immunized alpaca. The purity of the antigen and its correct conformation (biological activity) are very important for the immunization and later screening of suitable antibodies. Purity of the protein antigen is generally at least over 90%.
2. Separation of lymphocytes: cell separation in time can effectively prevent hemolysis after blood collection to achieve the best separation effect.
3. The immunization cycle can also affect the immunization effect. Based on our experiences, an interval of 1-2 weeks will induce a good immune response in most alpacas.
Recommended process for phage library construction:
1. RNA extraction: Use RNase-free materials and reagents for the RNA isolation. Dissolve the peripheral blood lymphocytes stored in Trizol on ice, transfer them to a 1.5 mL Eppendorf tube, add 1/5 volume of chloroform, vortex and mix; keep at room temperature for 5 minutes, then centrifuge at 12,000g for 15 minutes at 4°C; transfer the supernatant to a new Eppendorf tube; add an equal volume of isopropanol and mix; keep on ice for 30 minutes, then centrifuge at 12,000g at 4°C for 10 minutes; wash the precipitate with 75% ethanol, centrifugation at 7,500g for 5 minutes at 4°C, discard the supernatant, and air dry the pellet at room temperature and dissolve the pellet in an appropriate amount of RNase-free water. Measure RNA at 260nm and run RNA sample on a 1%(w/v) agarose gel for electrophoresis to evaluate the quality of total RNA.
2. Reverse transcription: according to the instructions of the reverse transcription kit, the total RNA obtained in the previous step will be divided into two equally, and reverse transcribed into cDNA. The primers used are Oligo dT and random primers, respectively. cDNA is relatively stable and can be stored at -80°C for later use.
3. Diverse VHH DNA fragments amplification: Amplify variable domains of all immunoglobulin heavy chains using reverse transcribed cDNA as template and perform PCR with Taq DNA Polymerase Hot Start enzyme. The PCR reaction system consists of 2 µL of cDNA template, 2 µL of Alpa001F primer, 2 µL of Alpa001R primer, 5 µL of 10x Taq Buffer, 4 µL of dNTP, 0.25 µL of Taq (HS), and ddH2O to a total volume of 50 µL. The PCR reaction cycle: 98°C for 3 minutes; 95°C for 30 seconds, 57°C for 30 seconds, 72°C for 40 seconds, increase by 2 seconds each cycle, and repeat 22 cycles; 72°C for 5 minutes. Run the PCR product on 1% agarose gel electrophoresis. PCR band of about 1000bp and band of about 700bp can be observed on the gel. Purify and recover DNA fragments of the 700bp band representing the heavy chain antibody repertoires. The gel-extracted and purified DNA fragments in the previous step are used as template using nested primers to re-amplify VHH open reading frame DNA fragments. The PCR reaction system is listed here: DNA template 2 µL, Alpa002F primer 2 µL, Alpa002R primer 2 µL, 10x Taq Buffer 5 µL, dNTP 4 µL, Taq (HS) 0.25 µL, ddH2O supplemented to 50 µL. The PCR reaction cycle: 98°C for 3 minutes; 95°C for 50 seconds, 55°C for 30 seconds, 72°C for 40 seconds, repeating 12 cycles; 72°C for 10 minutes. Purify and recover expected DNA band (about 400bp) using a PCR purification and recovery kit.
4. VHH cloning into phage plasmids: enzyme (SacI and SpeI in our case) digest and purify the VHH encoding fragments and phage vector separately, perform ligation reaction. Recover the ligation product using a DNA purification and recovery kit.
5. TG1 transformation: put sterile electroporation cuvettes on ice to pre-cool. Thaw 50 µL of TG1 competent cells, add 100 ng of the recovered ligation product, and transfer the mixed competent cells and ligation product to the pre-cooled electroporation cuvettes. Use transformation program for bacteria to electroporate. After electroporation, 1 mL of SOC medium will be added to the transformed competent cells. At least 20 electroporations will be performed, and the cells will be incubated at 37°C for 60 minutes. Put the cells on ampicillin-resistant LB plates for overnight growth. Scrape, rinse and collect the bacteria on plate next day with 2xYT medium, store at -80°C after adding 20% glycerol.
6. Rescue and amplification of phages from immune library: After mixing the scraped bacteria in the previous step, transfer about 10^9 bacteria to 100 mL of 2x YT medium with ampicillin, and incubate at 37°C and 220 rpm until OD600nm reaches 0.5. Add helper phage into bacteria at a ratio of 20:1(helper phage:bacteria), incubate at 37°C without shaking for 30 minutes. Add Kanamycin at a final concentration of 50 µg/mL, and incubate overnight at 30°C in a shaker (250rpm). Centrifuge the overnight bacteria culture at 13,000 rpm at 4°C for 5 minutes, transfer the supernatant to a new Eppendorf tube, add 1/4 volume of pre-cooled 5x PEG8000/NaCl, and incubate on ice for 30 minutes. Centrifuge at 13,000 rpm at 4°C for 10 minutes to remove the supernatant and add 1 mL of PBS buffer to dissolve the pellet. Add 250 µL of 5X PEG8000/NaCl again, incubate on ice for 10 minutes, centrifuge at 16,000 xg for 15 minutes at 4°C, remove the supernatant and dissolve the pellet in 1 mL of PBS to obtain a phage library, which can be stored at -80°C with 20% glycerol for long-term or at 4℃ for short-term (1-2 weeks).
Tips:
A library with larger capacity and better diversity is a pre-requisite for successful screening of nanobodies; The key points that affect the capacity and diversity of the library include: RNA degradation during RNA extraction, templates and primers during reverse transcription, primers design during amplification, PCR cycles, the size of the ligation system, and transformation efficiency of the bacteria.
Recommended procedures for single domain antibody screening and identification:
1. Antigen coating of the immunotube: add 50 µg of antigen to 2 mL of PBS (pH7.4) and then transfer to the immunotube, incubate overnight at 4°C with slow rotation.
2. Blocking: add amplified and purified phage to 1 mL of 3% BSA and incubate at room temperature with rotation for 2 hours. At the same time, add 2-3 mL of 3% BSA to the coated immunotube, and incubate with rotation at room temperature for 2 hours.
3. Incubating phage with antigen: Wash the immunotube with PBS containing 0.01% Tween20 3 times for 5 minutes each time. Add the blocked phage library to the blocked immunotube, add PBS to 2-3 mL, and incubate with rotation at room temperature for 1 hour.
4. Washing: Wash the immunotube with PBS containing 0.1% Tween20 20 times for 5 minutes each time.
5. Elution: Discard the liquid in the immunotube, remove the residual liquid as much as possible, add 1 mL 0.25 mg/mL Trypsin solution. Rotate at room temperature for 30 minutes and then add 10 µL of 10% AEBSF to inhibit the protease activity and terminate the elution, then transfer the phage-containing solution from the immunotube to a new 1.5 mL Eppendorf tube.
6. Eluted phage titer detection: Take 10 µL of the first round of phage eluate, dilute 10-fold gradient in a 1.5 mL Eppendorf tube with PBS, and dilute 10 to 1010 fold. Add 90 µL of TG1 bacterial culture at an OD600nm of 0.5-0.55 to each dilution Eppendorf tube, incubate at 37 °C for 30 minutes after mixing. Take 5µL bacterial culture of each dilution and drop onto 2×YT solid medium (supplemented with Ampicillin), air dry and then incubate overnight at 37 °C. Count the number of single colonies and then calculate the titer of the phage eluate by the number of single colonies with specific dilution.
7. Rescue and amplification of the first round of phage eluate: Take 500 µL of the phage eluate into 5 mL of bacterial culture at OD600 of 0.5-0.55, and continue to incubate at 37°C without shaking for 30 minutes. Spread all bacterial culture evenly on a culture plate containing 100 µg/mL Ampicillin containing 2% glucose, and culture at 37°C overnight. The next day, scrape,rinse and collect the colonies into a tube, which is the amplified bacterial sub-library. Repeat the screening procedure at least three times. The eluted phage can be sequenced by NGS to obtain a DNA sequence library of candidate nanobodies that may potentially can bind to the antigen.
8. Screening for antigen binders
- 8.1 The eluted phages from the last round of screening were serially diluted, add 100 µL of each dilution to the TG1 bacterial culture at OD600nm of 0.5, incubate at 37°C for 30 minutes without shaking, and put on a 2x YT culture plate containing ampicillin for overnight growth at 37°C.
- 8.2 Randomly pick 192 single colonies and inoculate each onto a 96-well cell culture plate containing 200µL 2x YT medium with ampicillin. It will be used as a seed plate after overnight culture at 37°C. Take 2 µL of each bacterial culture from seed plate and add it to a new 96-well plate (each well contains 200 µL of 2x YT medium, 100 µg/mL ampicillin). After 5 hours of incubation at 37°C, add helper phages to the culture wells (helper phage M13K07) with a bacteria/phage ratio of 1:20 at 37°C for 30 minutes, then add kanamycin with a final concentration of 50 µg/mL, and culture at 30°C overnight. Centrifuge the bacterial culture next day to obtain the supernatant containing phage. Stored at 4°C for the following use.
- 8.3 Coat an ELISA plate with the antigen (1 ng/µL, the coating solution is PBS pH7.4, 100 µL/well). At the same time, coat BSA as a control in parallel overnight at 4°C; discard the liquid in the ELISA plate, add 200 µL of PBS to each well, wash the ELISA plate 3 times at room temperature for 10 minutes each time; add 200 µL of blocking solution (3% BSA) to each well at room temperature for 1 hour.
- 8.4 Discard the blocking solution, add 200 µL of PBST (1×PBS with 0.1% Tween20) buffer to each well, and wash the ELISA plate at room temperature 3 times for 10 minutes each time.
- 8.5 Add 120 µL of 3% BSA to each well, then add 80 µL of the phage supernatant obtained in step 8.2, and incubate at room temperature for 2 hours; discard the liquid in the ELISA plate, and add 200 µL of PBST buffer to each well to wash 3 times. 10 minutes each time.
- 8.6 Add M13Bacteriophage Antibody (HRP), Mouse Mab, 0.1 ng/µL diluted in blocking solution to each well, 100 µL/well, incubate at room temperature for 1 hour.
- 8.7 Discard the liquid in the ELISA plate, add 200 µl PBST buffer to each well and wash 3 times for 10 minutes each time.
- 8.8 Add 100 µL of TMB single-component chromogenic solution to each well, and put the plate in the dark for 2-3 minutes for color development. Add 100 µL of 1M HCl to each well to terminate, read the OD450nm value with a microplate reader, record and save the result.
- 8.9 Select colonies with a large ratio of absorbance values in the antigen-coated wells to the corresponding control wells after two independent phage-Elisa analyses for colony sequence analysis to obtain the VHH encoding frame sequence.
Tips:
1. The amount of antigen for coating is related to the molecular weight, hydrophilicity, structure of the antigen, as well as the choice of coating buffer and coating medium. Appropriate coating is the basis for successful screening. It is recommended to carry out preliminary experiments to determine the coating conditions or select the method of screening such as magnetic beads.
2. Determine the titer of phage after elution. After 2-4 rounds of antigen-coated panning, the enrichment of phage should be within a reasonable range.

Phage eluate evaluation
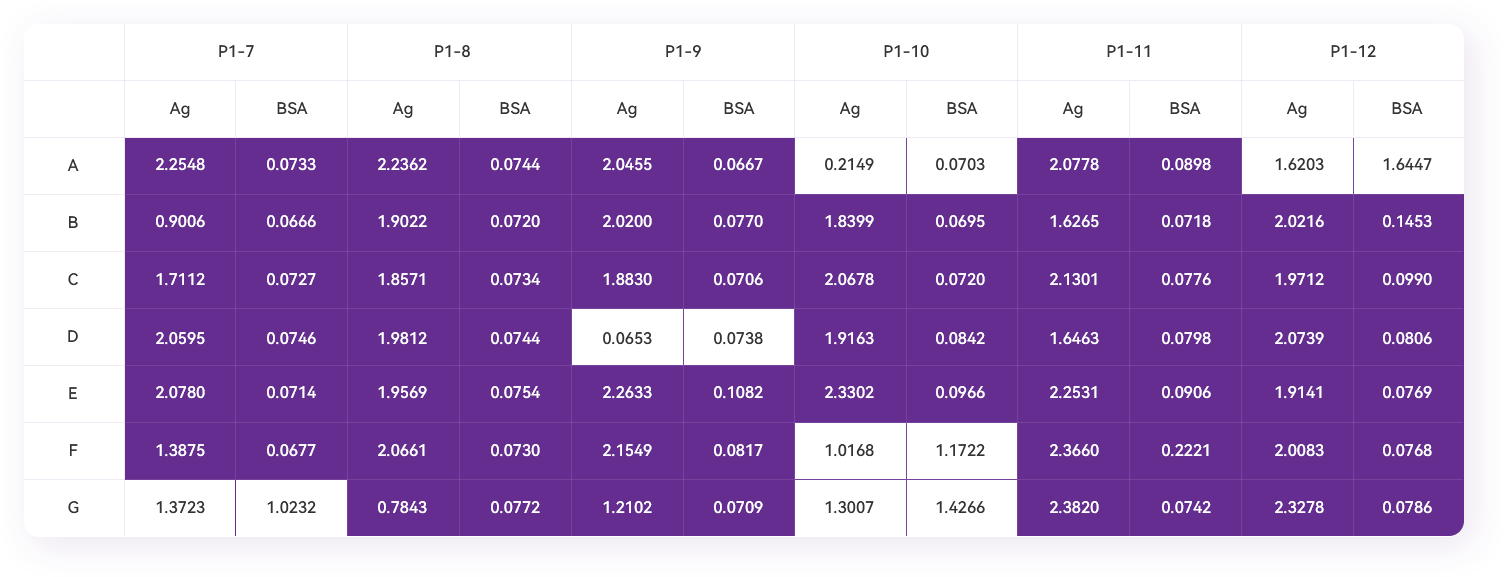
Phage Elisa
Methods for single domain antibody expression and purification:
VHH candidates can be expressed in E. coli, yeast or mammalian cell lines. In order to verify whether they can bind to antigens specifically, plasmids can be constructed for rapid expression and purification in E. coli or yeast strains.
Induced expression and purification of Nanobodies in E. coli:
After the plasmid construction sequence is confirmed, transform the expression strain, such as BL21, pick a single colony for culture, induced expression with IPTG, and purify the soluble protein in the supernatant after the cell is lysed and centrifuged, and affinity chromatography, ion exchange chromatography and size-exclusion chromatography can be used for purification.
Expression and purification of nanobodies in yeast such as P. pastoris:
After the plasmid construction sequence is confirmed, electroporate yeast such as GS115 or X33 strain with linearized plasmid. Choose several single colonies for methanol-induced expression. purify nanobodies from medium supernatant.
The advantage of E. coli expression is that it can be used for quick expression, but the disadvantage is that some nanobodies can form inclusion bodies, and the intracellular reducing environment is not conducive to the formation of disulfide bonds in nanobodies. The advantage of Pichia expression system is that the expressed VHH can be secreted outside of the yeast cell. The purity of the VHH in the medium supernatant is relatively high to make the purification easier, and the extracellular oxidative environment is conducive to the formation of disulfide bond in the nanobodies, which is crucial to the stability of nanobodies.
Affinity assay:
The equipment used for affinity assay is BiacoreT200. For the specific operation process, please refer to the "BiacoreT200 for Protein and Protein Binding Operation Guide".